1. 시스템 및 소프트웨어 환경
1.1 시스템은 x86기반 리눅스 클러스터로, MPI용 네트웍은 이더넷이며,
운영체제는 CentOS 4.2 (RedHat EL4 update2 호환) 입니다.
1.2 vmd를 실행하는 pc는 windows xp환경이며, 이외에도 여러가지 운영체제를
지원합니다.
1.3 패키지 버전
- Namd 2.5 Source ( http://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD
: 오픈소스 )
- VMD 1.8.5 ( http://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD
: 오픈소스 )
* namd및 mpich설치는 이전 문서를 참조 바랍니다.
2. vmd 1.8.5 설치
- 다운로드한 vmd185.exe를 더블클릭하여 설치 합니다.
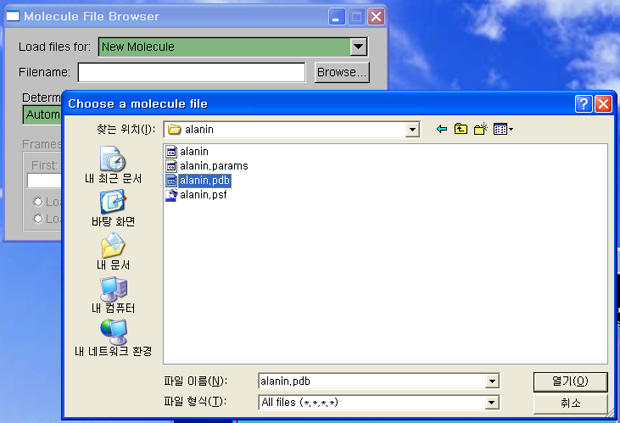
3. 실행용 예제(alanin)의 구조, 파라메터, 좌표 파일들을 클러스터로 부터,
다운로드하여, pc에 저장합니다.
(alanin.params, alanin.pdb, alanin.psf)
* vmd가 한글을 읽지 못하므로, 한글 윈도우의 경우는 [바탕화면]과 같은
경로에 파일을 위치시키면 안됩니다.
ex>
==========================================================================
D:\>dir d:\alanin
D 드라이브의 볼륨에는 이름이 없습니다.
볼륨 일련 번호: 2400-08F9
d:\alanin 디렉터리
2006-10-04 오후 08:26 .
2006-10-04 오후 08:26 ..
2006-10-04 오후 07:37 517 alanin
2006-10-04 오후 07:37 17,389 alanin.params
2006-10-04 오후 07:37 5,748 alanin.pdb
2006-10-04 오후 07:37 12,953 alanin.psf
2006-10-04 오후 08:26 16,247 imd.vmd
5개 파일 52,854 바이트
2개 디렉터리 108,966,461,440 바이트 남음
==========================================================================
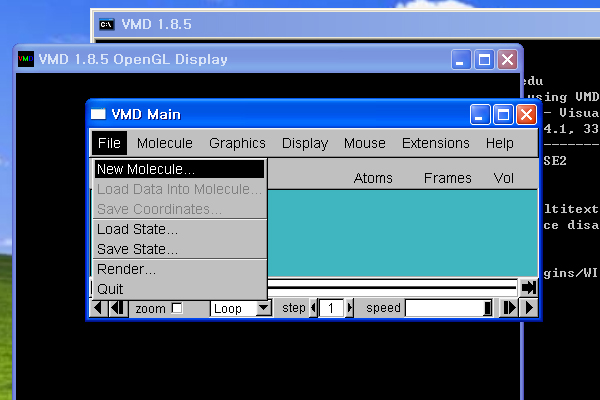
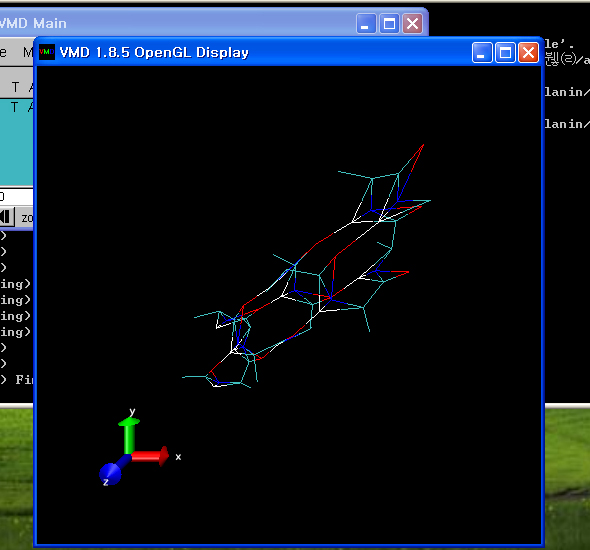
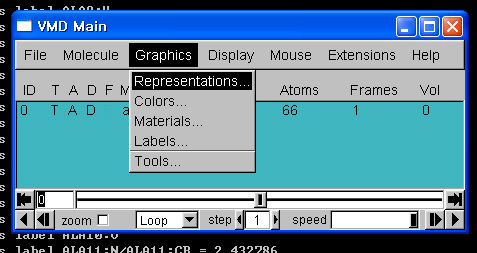
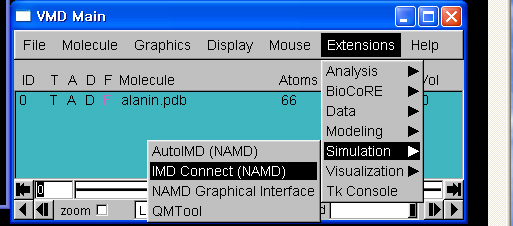
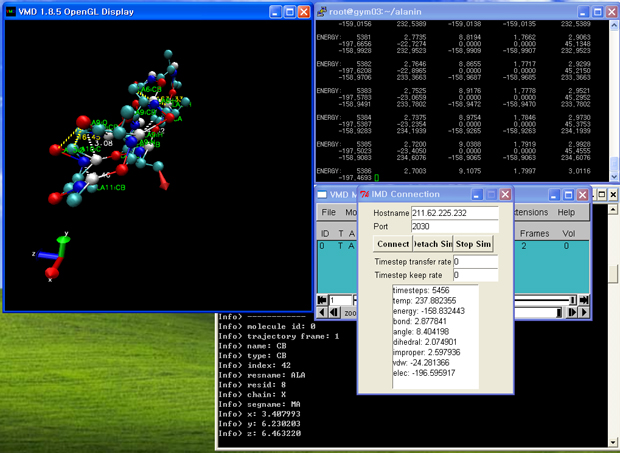
4. vmd 실행
- [시작]->[프로그램]->[VMD 1.8.5]를 클릭하여, vmd를 실행합니다.
|
5. namd 실행
- ssh터미널을 열고 마스터 노드에 로그인 하여, alanin예제가 있는 디렉토리로
이동하고, alanin파일에 IMD를 사용하도록 다음과 같은 설정을 추가합니다.
* 이때, numstep의 값을 충분히 주어야 imd가 빨리 끝나는 것을 방지 할 수
있습니다.
==========================================================================
# cd ~/alanin
# vi ./alanin
-------------------------------------------------------------
# This is a test namd configuration file
timestep 0.5
numsteps 50000
structure alanin.psf
parameters alanin.params
coordinates alanin.pdb
exclude scaled1-4
1-4scaling 0.4
outputname output
margin 1.0
stepspercycle 3
temperature 0
switching on
switchdist 7.0
cutoff 8.0
pairlistdist 9.0
#dcdfile alanin.dcd
#dcdfreq 10
#restartname alanin.restart
#restartfreq 10
#langevin on
#langevinTemp 300.0
#langevincol O
#constraints on
#fma on
#seed 791064881
IMDon yes
IMDport 2030
IMDfreq 1
IMDwait on
-------------------------------------------------------------
==========================================================================
- 이제 namd을 실행합니다.
==========================================================================
# vi ~/mf
--------------------------------------------
node001
node002
--------------------------------------------
# mpirun -np 2 -machinefile ~/mf -nolocal /usr/local/namd/Linux-i686-MPI/namd2 ./alanin
==========================================================================
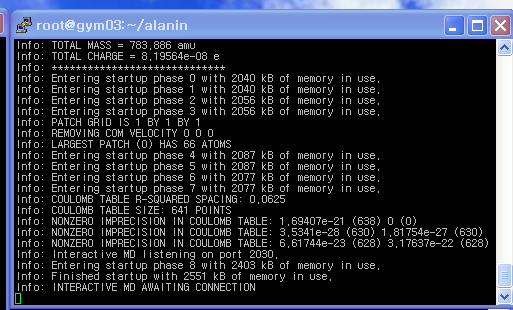
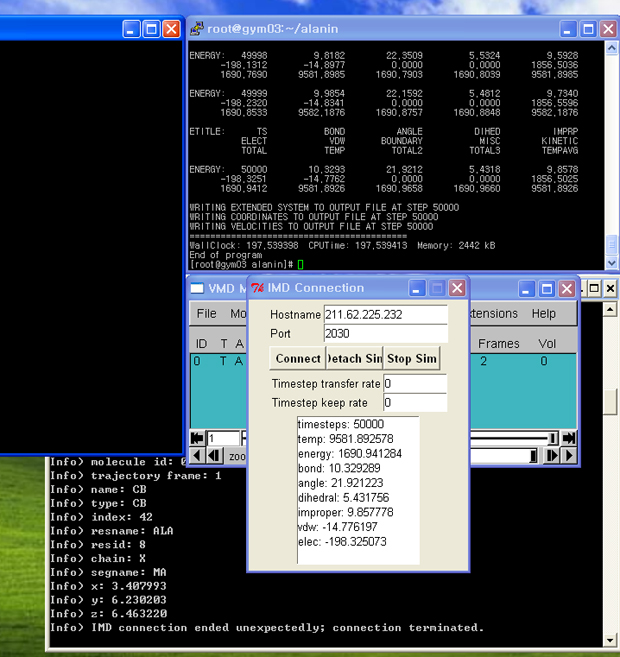
- namd가 실행되면 다음과 같이 imd접속을 기다린다는 메시지가 나옵니다.
|